An overview of the guidelines for caring for those affected
Rare diseases
New Guideline on Allan-Herndon-Dudley Syndrome
An international team has published a new guideline for diagnosing and treating the rare genetic disease. It serves as a tool for everyone who treats and cares for patients affected by it.
Allan-Herndon-Dudley syndrome is a rare congenital disease of the thyroid metabolism. It occurs in approximately one out of every 70,000 male newborns and its effects are devastating. Since early 2025, a medication has been approved in Europe that can alleviate symptoms and extend patients’ life expectancy. However, the dosage is complicated. A team of specialists working with Professor Johannes W. Dietrich from the Department of Diabetology, Endocrinology, and Metabolism at the Ruhr University Bochum St. Josef Hospital, Germany – and director of the Center for Rare Endocrine Diseases at the Center for Rare Diseases Ruhr (CeSER) – has published a guideline for treating the disease in the journal Hormone Research in Pediatrics from April 2, 2026.
Severe symptoms and short life expectancy
In Allen-Herndon-Dudley syndrome, a mutation of the MCT8 protein severely disrupts the transport of thyroid hormones into and out of certain cell types. The disease is inherited through the X chromosome and is very rare, and has devastating consequences for individuals afflicted by it: Severe developmental disorders with consequent major intellectual disability, muscle weakness, and cardiac arrhythmias are typical. “Patients usually never learn how to speak or walk, and, due to an elevated concentration of the thyroid hormone T3, they usually die prematurely from cardiovascular complications,” says Dietrich. “The average life expectancy is 35 years.”
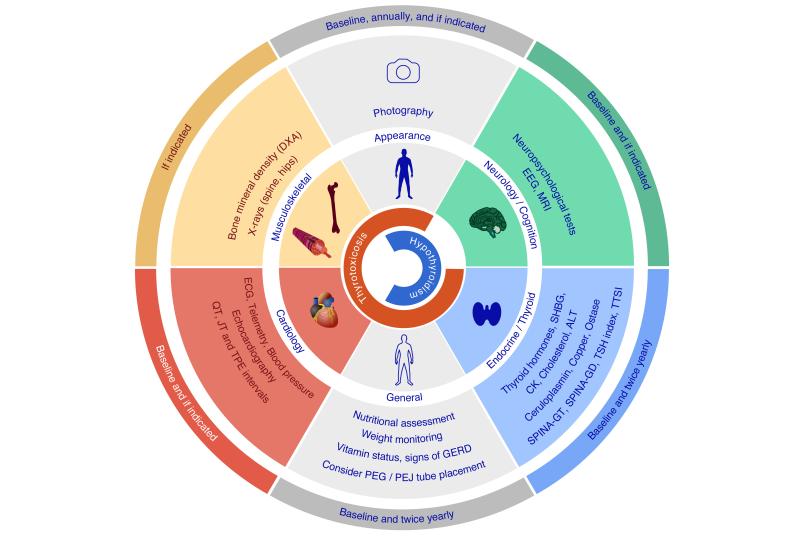
Individual treatment plan
A newly approved medication, the non-classical thyroid hormone TRIAC, is now available in Europe. This therapy can alleviate some of the symptoms and extend patients’ life expectancy, although the dosage is highly complex. A working group from Germany and Switzerland has developed the new guideline based on extensive research of the literature and their own clinical experience in pediatric neurology and endocrinology. It provides recommendations for a comprehensive, multidisciplinary treatment strategy for patients in all age groups while also considering the monitoring of the most important systems and secondary diseases. “Because the symptoms and long-term courses associated with this disease can vary widely, individual treatment plans for the patients are necessary,” says Dietrich.