Die Leitlinie zur Versorgung Betroffener im Überblick
Seltene Erkrankungen
Neue Leitlinie zum Allan-Herndon-Dudley-Syndrom
Ein internationales Team hat eine neue Leitlinie zur Diagnostik und Behandlung der seltenen genetischen Erkrankung veröffentlicht. Sie gibt Orientierung für alle, die Betroffene behandeln und pflegen.
Das Allan-Herndon-Dudley-Syndrom ist eine seltene angeborene Erkrankung des Schilddrüsenstoffwechsels. Sie tritt bei ungefähr einem von 70.000 männlichen Neugeborenen auf und hat katastrophale Folgen für die Betroffenen. Seit Anfang 2025 ist in Europa ein Medikament zugelassen, das Symptome lindern und die Lebensdauer verlängern kann. Die Dosierung ist allerdings kompliziert. Ein Spezialistenteam mit Prof. Dr. Johannes W. Dietrich, Sektion Diabetologie, Endokrinologie und Stoffwechsel am Universitätsklinikum der Ruhr-Universität Bochum St. Josef-Hospital und Leiter des Zentrums für Seltene Endokrine Erkrankungen am Centrum für Seltene Erkrankungen CeSER, hat eine Leitlinie zur Behandlung der Erkrankung im Journal Hormone Research in Pediatrics vom 2. April 2026 veröffentlicht.
Schwere Symptome und kurze Lebenserwartung
Beim Allan-Herndon-Dudley-Syndrom ist durch eine Mutation des Proteins MCT8 der Transport von Schilddrüsenhormonen in und aus bestimmten Zelltypen schwer gestört. Die Erkrankung ist X-chromosomal vererbt und sehr selten, geht aber für die Betroffenen mit katastrophalen Folgen einher: Schwere Entwicklungsstörungen mit konsekutiv schwerem Schwachsinn, Muskelschwäche und Herzrhythmusstörungen sind typisch. „Meist erlernen die Patienten weder zu sprechen noch zu gehen, und sie sterben durch eine erhöhte Konzentration des Schilddrüsenhormons T3 meist früh an Herz-Kreislauf-Komplikationen“, beschreibt Johannes W. Dietrich. „Die durchschnittliche Lebenserwartung liegt bei 35 Jahren.“
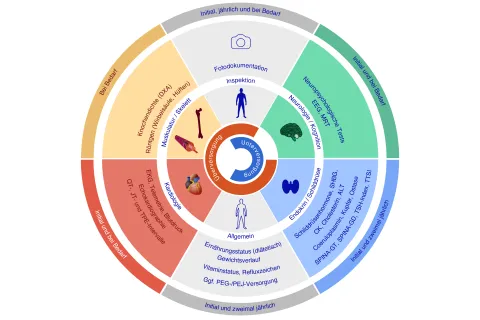
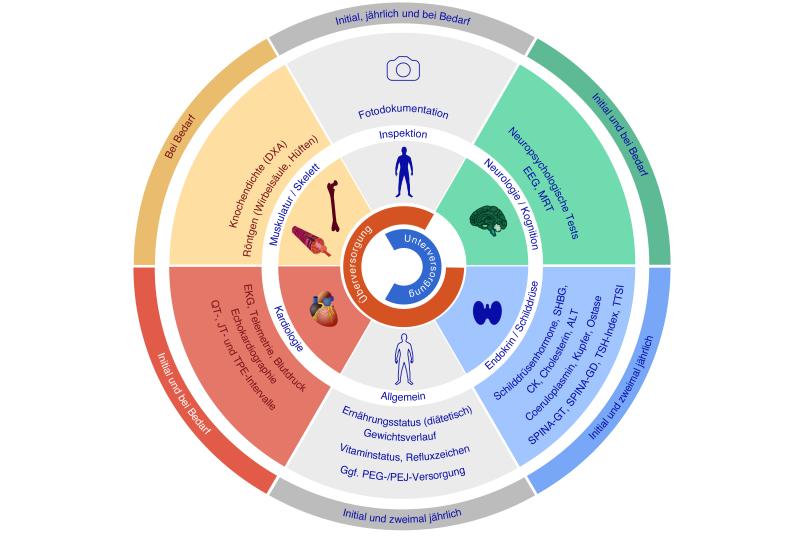
Individueller Behandlungsplan
Inzwischen gibt es mit dem nicht-klassischen Schilddrüsenhormon TRIAC ein neu zugelassenes Medikament in Europa. Die Therapie kann einen Teil der Symptome lindern und die Lebensdauer verlängern. Allerdings ist die Dosierung sehr schwierig. Eine Arbeitsgruppe aus Deutschland und der Schweiz hat auf Basis einer aufwändigen Literaturrecherche und eigener klinischer Erfahrungen aus der pädiatrischen Neurologie und Endokrinologie die Leitlinie entwickelt. Sie gibt Empfehlungen für eine umfassende, multidisziplinäre Behandlungsstrategie für Patienten aller Altersgruppen. Sie berücksichtigt dabei die Überwachung der wichtigsten Symptome und Folgeerkrankungen. „Weil die Symptome und Langzeitverläufe im Zusammenhang mit dieser Erkrankung sehr unterschiedlich sein können, sind individuelle Behandlungspläne für die Patienten notwendig“, so Dietrich.