
Medizin
Seltene Muskelkrankheit entdeckt
Nach der Ursache für diese Krankheit mussten die Bochumer Mediziner gründlich suchen. Und auch jetzt sind noch viele Fragen zu den zugrunde liegenden Mechanismen offen.
Muskelschwäche in den Beinen, ein unsicherer Gang, die ständige Gefahr zu stolpern – solche Symptome sind üblich für Menschen mit Muskelerkrankungen. Auf den Patienten, der mit diesen Beschwerden ins Universitätsklinikum Bergmannsheil kam, passte aber keine der bekannten Diagnosen. Eine ausführliche Untersuchung durch die Bochumer Mediziner ergab schließlich, dass sie es mit einer neuen Krankheit zu tun hatten.
„Wir haben sehr viel Zusatzdiagnostik gemacht“, erzählt Prof. Dr. Matthias Vorgerd von der Neurologischen Klinik Bergmannsheil, „aber wir konnten das verantwortliche Gen oder Protein am Anfang nicht eingrenzen.“ Da weitere Familienmitglieder des Patienten betroffen waren, gingen die Ärzte von einer vererbbaren Erkrankung aus; sie stießen mit Privatdozentin Dr. Sabine Hoffjan von der Humangenetik der RUB detailliertere genetische Analysen an – und wurden fündig. Das Gen BICD2 war bei allen Betroffenen verändert. Die Ursache bestätigte sich später in einer zweiten erkrankten Familie.
Bekannter Krankheitsauslöser
BICD2 war zuvor schon als Krankheitsauslöser bekannt gewesen – allerdings hatte noch niemand ein BICD2-Syndrom beschrieben, das sich in einer veränderten Skelettmuskulatur geäußert hatte. Stets war das Problem vom Nervensystem ausgegangen. Nun sahen die Mediziner einen deutlichen Befall der Unter- und Oberschenkelmuskulatur, während im Nervensystem keine relevanten Veränderungen erkennbar waren. Dieser neuen Muskelkrankheit wollte Matthias Vorgerd genauer auf den Grund gehen. „Die Krankheit möglichst gut zu beschreiben ist wichtig, um Aussagen zum Vererbungsgang, zum Verlauf und zu Therapieoptionen machen zu können“, erklärt der Neurologe. Er zog die Arbeitsgruppe von Prof. Dr. Wolfgang Linke vom RUB-Institut für Physiologie hinzu.

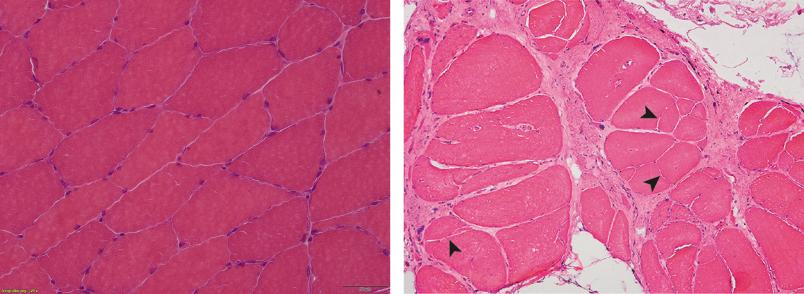
Die Physiologen um Dr. Andreas Unger analysierten die Zellen aus den Biopsien betroffener Patienten mit verschiedenen Labortests. Unter anderem fertigten sie hochauflösende elektronenmikroskopische Aufnahmen an. Sie offenbarten starke Veränderungen in der normalerweise sehr geordneten Muskelstruktur. Myofibrillen, die Bausteine der Muskelfasern, waren degeneriert. Aber auch manche Zellorganellen waren von der Krankheit betroffen. Die Mitochondrien, die Kraftwerke der Zellen, hatten eine andere Form als im gesunden Zustand, und der Golgi-Apparat, eine Art Poststelle für Proteine, war größer und zerklüfteter als normalerweise.
Schwarzes Loch der Physiologie
Aber wie löst der Defekt im Gen BICD2 all diese Veränderungen in der Zelle aus? „Es ist noch zu früh für eine eindeutige Antwort“, sagt Andreas Unger. Klar ist, dass das Gen einen Bauplan für ein Protein enthält, das entscheidend für zelluläre Transportprozesse ist. „Transportprozesse in Muskelzellen sind so eine Art Schwarzes Loch, man weiß bislang nur sehr wenig über sie“, so Unger.
Soll eine Fracht innerhalb der Zelle transportiert werden, wird sie dazu in ein Membranbläschen verpackt und von einem beweglichen Protein befördert. Das BICD2-Protein fungiert als Adapter zwischen der zu transportierenden Fracht und dem Motorprotein. Es bestimmt mit, welche Fracht transportiert wird und wohin, und es reguliert die Geschwindigkeit des Motorproteins. Vieles über die physiologische Funktion von BICD2 ist aber noch nicht erforscht, etwa welche Fracht es überhaupt transportiert. Daher kann Andreas Unger bislang nur spekulieren, wie sich der Gendefekt auf die Transportprozesse auswirkt. „Die dreidimensionale Struktur des BICD2-Proteins ist bekannt, es sieht aus wie ein dreiteiliges Klappmesser“, beschreibt er. „Um die Fracht aufzunehmen, muss es entfaltet werden. Denkbar wäre, dass das Protein aufgrund der Mutation nicht mehr richtig aufklappt. Das ist aber nur eine Hypothese.“
Gestörte Regeneration
Bekannt ist: Wenn das krankhaft veränderte BICD2-Gen in ein Protein übersetzt wird, wird aufgrund des Fehlers im Erbgut eine einzelne falsche Aminosäure eingebaut. Das reicht offenbar aus, um die Funktion des Proteins stark zu beeinträchtigen. Die Mutation wird dominant vererbt; wer auf einem Chromosom im Chromosomenpaar eine krankhafte Anlage besitzt, erkrankt.

Im weitesten Sinne ist es wie mit Lebensmitteln, die sich nicht ewig halten.
Andreas Unger
Basierend auf den bisherigen Untersuchungen nehmen Andreas Unger und seine Kollegen an, dass das kranke BICD2-Protein den gerichteten Transport in Muskelzellen beeinträchtigt; er ist wichtig für die Regeneration der Zellmembran. Diese ist durchsetzt von Proteinen, die regelmäßig erneuert werden müssen – und dafür müssen die Ersatzproteine zur Zellmembran gelangen. „Im weitesten Sinne ist es wie mit Lebensmitteln, die sich nicht ewig halten“, vergleicht Unger. „Auch die Membranproteine brauchen einen Wechsel, und der scheint bei BICD2-Patienten gestört zu sein.“
Breites Krankheitsspektrum
Neue Befunde deuten darauf hin, dass die BICD2-Muskelschwäche kein klar abgegrenztes Syndrom ist, sondern dass es ein ganzes Spektrum von BICD2-ausgelösten Krankheiten gibt. „Wahrscheinlich gibt es eine schwere Krankheitsform, die schon bei der Geburt ausgeprägt ist und zum Tod in den ersten Lebensmonaten führt“, sagt Matthias Vorgerd. Einen entsprechenden Fall untersucht das Team gerade.
Diese Symptomatik steht an dem einen Ende des klinischen Spektrums der BICD2-Krankheiten; am anderen Ende stehen Ausprägungen mit relativ milden Beschwerden, die erst im hohen Erwachsenenalter zutage treten. Dieses BICD2-Spektrum umfasst Krankheiten, deren Ursache entweder nur in der Skelettmuskulatur liegt oder nur im Nervensystem, aber auch Krankheiten, die eine Mischform aus beidem sind.

Mit ihrer ersten Beschreibung der muskulären BICD2-Krankheit haben Vorgerd, Unger und Kollegen andere Kliniker für das Krankheitsspektrum sensibilisiert. „Wenn ein Arzt solche Krankheitsbilder sieht und ein Vererbungsmuster dahinter vermutet, muss er die Familien künftig auf BICD2-Defekte untersuchen“, erklärt Matthias Vorgerd. „Kliniker sollen nun einen Diagnoseschlüssel in die Hand bekommen, mit dem sie das gezielt tun können.“
Fernziel: Therapie
Gemeinsam mit anderen Arbeitsgruppen in Deutschland wollen die Bochumer Wissenschaftler die BICD2-Krankheit in den kommenden Jahren besser verstehen. Dafür werden auch neue Einblicke in die Transportprozesse in gesunden Muskelzellen notwendig sein. „Später könnte uns dieses Wissen helfen, über Therapieoptionen nachzudenken“, prognostiziert Unger. „Denkbar wäre zum Beispiel, dass man das nicht richtig transportierte Molekül durch ein anderes ersetzen kann oder auf ein anderes Frachtsystem umstellen kann“, überlegt der Wissenschaftler. Das geht aber nur, wenn man weiß, wie die Prozesse im gesunden Zustand sein sollten.
Das sind langwierige Prozesse, für die es im Moment überhaupt kein Geld gibt.
Matthias Vorgerd
Bis eine Therapie vorliegt, werden noch Jahre verstreichen. Mindestens zehn, schätzt Matthias Vorgerd. Selbst wenn es eine Therapieidee gäbe, müsste diese erst in der Zellkultur, dann an Tieren getestet werden. „Das sind langwierige Prozesse, für die es im Moment überhaupt kein Geld gibt“, erklärt der Neurologe. Die BICD2-Defekte sind seltene Krankheiten. Nicht leicht sei es, dafür an Fördergelder zu kommen. Die bisherigen Arbeiten wurden von der Heimer-Stiftung unterstützt. Für die Industrie sei diese Forschung nicht rentabel.
„In Deutschland gibt es derzeit vielleicht 10 bis 15 BICD2-Patienten“, weiß Vorgerd. Somit ist auch das Material für die Forschung stark begrenzt. Dennoch beobachten die Bochumer eine positive Entwicklung: „Unter Fachkollegen spricht sich herum, dass wir uns in Bochum auf Fälle mit muskulärer Ursache spezialisiert haben“, erklärt Andreas Unger. So hätten Kollegen ihnen bereits Gewebeproben weiterer Fälle zukommen lassen – und das bringt die BICD2-Forschung voran.






