Bioinformatik
Künstliche Intelligenz faltet RNA-Moleküle
Die räumliche Struktur von RNA-Molekülen ist entscheidend für ihre Funktion. Sie vorherzusagen ist aber eine Herausforderung.
Für die Funktion vieler Biomoleküle ist ihre dreidimensionale Struktur entscheidend. Daher sind Forschende nicht nur an der Sequenz der Einzelbausteine von Biomolekülen interessiert, sondern auch an ihrer räumlichen Struktur. Mithilfe von Künstlicher Intelligenz (KI) können Bioinformatikerinnen und -informatiker bereits zuverlässig die dreidimensionale Struktur eines Proteins aus dessen Aminosäuresequenz vorhersagen. Für RNA-Moleküle steht diese Technik jedoch erst am Anfang. Forschende der Ruhr-Universität Bochum (RUB) beschreiben in der Zeitschrift „PLOS Computational Biology“ vom 7. Juli 2022 einen Weg, um mit KI die Struktur bestimmter RNA-Moleküle zuverlässig aus ihrer Nukleotidsequenz vorherzusagen.
Für die Arbeiten kooperierten die Teams um Vivian Brandenburg und Prof. Dr. Franz Narberhaus vom Lehrstuhl für Biologie der Mikroorganismen der RUB mit Prof. Dr. Axel Mosig vom Kompetenzbereich Bioinformatik des Bochumer Zentrums für Proteindiagnostik.
Zellumgebung muss berücksichtigt werden
„Oft wird RNA nur als Bote zwischen der genomischen DNA und den Proteinen verstanden“, sagt Axel Mosig. „Aber viele RNA-Moleküle übernehmen zelluläre Funktionen.“ Wichtig dafür ist ihre räumliche Struktur. Ähnliche Bereiche in einer Nukleotidsequenz können sich zusammenlagern und dadurch dreidimensionale Anordnungen bilden.
„Diese Selbstähnlichkeiten in einer RNA-Sequenz zu identifizieren ist wie ein mathematisches Puzzle“, erklärt Vivian Brandenburg. Für dieses Puzzle gibt es ein biophysikalisches Modell mit entsprechenden Vorhersage-Algorithmen. Das Modell kann allerdings nicht die zelluläre Umgebung der RNA berücksichtigen – und diese beeinflusst den Faltungsprozess ebenfalls. „Würde die RNA isoliert in wässriger Lösung schwimmen, könnte das Modell die Struktur sehr präzise vorhersagen“, so Brandenburg. Aber in einer lebenden Zelle sind viele andere Bestandteile enthalten.
Hier kommt die Künstliche Intelligenz ins Spiel. Diese kann anhand von bekannten Strukturen subtile Muster lernen, die sich aus der zellulären Umgebung ergeben. Diese Erkenntnisse könnte die KI dann in ihre Strukturvorhersagen einbeziehen. Für den Lernprozess braucht die KI aber ausreichend Trainingsdaten – und die fehlen in der Praxis eigentlich.
Trainingsdaten mit einem Trick gewinnen
Um das Problem der fehlenden Trainingsdaten zu lösen, bediente sich das Bochumer Team eines Tricks: Die Forschenden arbeiteten mit bekannten RNA-Struktur-Motiven. Mit einer Art Rückwärtsgang konnten sie aus den Energiemodellen dieser Strukturen fast beliebig viele Nukleotidsequenzen generieren, die in diese räumlichen Strukturen falten würden. Mithilfe dieses sogenannten inversen Faltens erzeugten die Wissenschaftlerinnen und Wissenschaftler viele Paare aus Nukleotidsequenzen und Strukturen, mit denen sie die KI trainieren konnten.
Neue Strukturen zuverlässig vorhersagbar
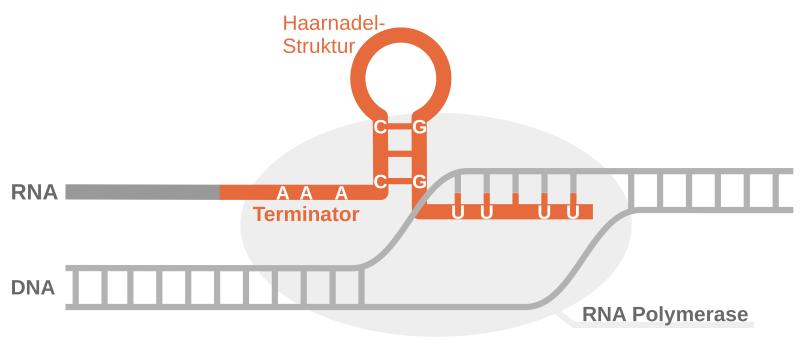
Anschließend konfrontierten die Forschenden die KI mit einer neuen Aufgabe: Sie musste die Struktur von bestimmten bakteriellen RNA-Molekülen vorhersagen. Diese Moleküle – Transkriptions-Terminatoren genannt – sind in Bakterien wichtige Stopp-Signale bei der Übersetzung genomischer DNA. Oft sind sie, wie viele andere RNA-Moleküle mit wichtigen zellulären Funktionen, im Erbgut versteckt und schwierig von Bereichen mit anderen Funktionen zu unterscheiden.
Die Künstliche Intelligenz konnte die typische Struktur der Transkriptions-Terminatoren, die an eine Haarnadel erinnert, zuverlässig erkennen und vorhersagen. Das konnte das Forschungsteam anhand von öffentlich zugänglichen experimentellen Daten nachweisen.
„Während KI-Ansätze bei der Vorhersage von Proteinstrukturen mittlerweile fast unumgänglich sind, steht die Entwicklung bei RNA-Strukturen erst am Anfang“, fasst Axel Mosig zusammen.